Ensemble-PlantSCL: Ensemble of multiple classifiers for multilabel
classification of
plant protein subcellular localization
Abstract: The accurate prediction of protein localization is a critical
step in any functional genome annotation process. This paper proposes an
improved strategy for protein subcellular localization prediction in plants
based on multiple classifiers to improve prediction results in terms of both
accuracy and reliability. The prediction of plant protein subcellular
localization is challenging because the underlying problem is not only a
multiclass, but also a multilabel problem. Generally, plant proteins can be
found in 10-14 locations/compartments. The number of
proteins in some compartments (nucleus, cytoplasm, and mitochondria) is
generally much greater than that in other compartments (vacuole, peroxisome,
Golgi, and cell wall). Therefore, the problem of imbalanced data usually
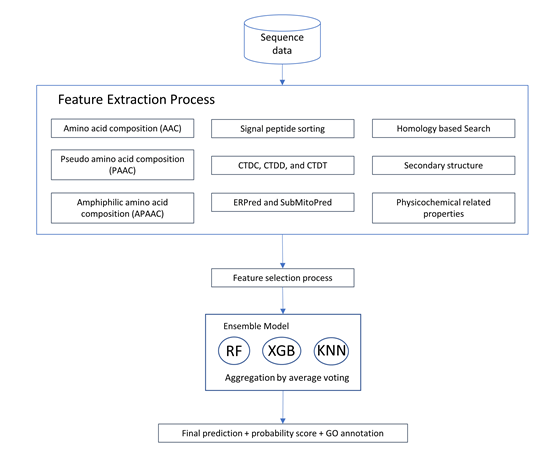
arises. Therefore, we propose an ensemble machine learning method based on
average voting among heterogeneous classifiers. We first extracted various
types of features suitable for each type of protein localization to form a
total of 479 feature spaces. Then, feature selection methods were used to
reduce the dimensions of the features into smaller informative feature subsets.
This reduced feature subset was then used to train/build 3 different individual
models. In the process of combining the 3 distinct classifier models, we used
an average voting approach to combine the results of these 3 different
classifiers that we constructed to return the final probability prediction. The
method can predict subcellular localizations in both single- and multilabel
locations based on the voting probability. Experimental results indicated that
the proposed ensemble method can achieve correct classification with an overall
accuracy of 84.58% for 11 compartments on the basis of
the testing dataset.
Please cite:
Wattanapornprom, W., Thammarongtham, C., Hongsthong,
A., Lertampaiporn, S. Ensemble of multiple
classifiers for multilabel classification of plant protein subcellular
localization.2021. Life 11 (4), 293.
Download Link: Training and Testing data, Standalone Program
Command: perl EnsemblePlantSCL.pl input_fasta_file
(mode1: probability for predicted location only)
perl EnsemblePlantSCL_distribution.pl input_fasta_file
(mode2: probability distribution for all locations)
***** Fasta seq must has length > 10 aa and contains
only standard amino acids *****
###Example:
perl EnsemblePlantSCL_distribution.pl ./Seq_Data/pln_tag_EMR.fa
###Output:
############
Prediction results by EnsemblePlantSCL
(mode2) ############
(Probability distribution of 1:Cell
membrane,2:Cell
wall,3:Cytoplasm,4:ER,5:Extra,6:Golgi,7:mitochondria,8:Nucleus,9:Peroxisome,10:Plastid,11:Vacuole)
Seq.1 predicted as 4:ER - with prob. dist:
0.035,0.005,0.253,*0.588,0.007,0.007,0.043,0.01,0.001,0.044,0.007
Seq.2 predicted as 4:ER - with prob. dist:
0.032,0,0.161,*0.401,0.004,0.012,0.214,0.144,0.002,0.02,0.01
Seq.3 predicted as 4:ER - with prob. dist:
0.041,0.001,0.344,*0.45,0.028,0.003,0.042,0.05,0.003,0.031,0.007
Seq.4 predicted as 4:ER - with prob. dist:
0.067,0.002,0.131,*0.4,0.015,0.035,0.203,0.083,0.004,0.042,0.018
Seq.5 predicted as 4:ER - with prob. dist:
0.036,0,0.142,*0.408,0.003,0.031,0.314,0.059,0,0.004,0.002